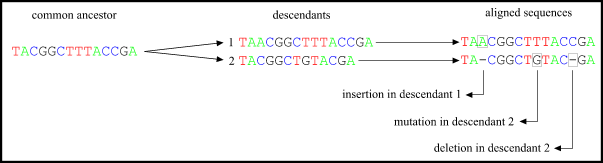
Example : two sequences :
TCAGACGATTG
TCGGAGCTG
How can we get the best alignment ? There are several possibilities :
1. Reduce the number of mismatches :
TCAG-ACG-ATTG
|| | | | | | 0 mismatches 7 matches 6 gaps
TC-GGA-GC-T-G
2. Reduce the number of gaps :
TCAGACGATTG
|| || 5 mismatches 4 matches 2 gaps
TCGGAGCTG--
3. Reduce neither the number of gaps nor the number of mismatches :
TCAG-ACGATTG
|| | | | | 2 mismatches 6 matches 4 gaps
TC-GGA-GCTG-
4. Same as 3. but one base (or gap) moved :
TCAG-ACGATTG
|| | | | | | 1 mismatch 7 matches 4 gaps
TC-GGA-GCT-G
Which of these is now the best alignment ??
There are several alignment algorithms to choose the best alignment. Let's use a simple one in this example :
D = y + sum(wkzk)
with :
D = distance
y : number of mismatches
w : penalty for gaps of length k
z : number of gaps of length k
Take gap penalty for gap length 1 = 2
Take gap penalty for gap length 2 = 6 (short gaps occur more frequent than long gaps)
in 1. : 0 + {(2 x 6) + (6 x 0)} = 12
in 2. : 5 + {(2 x 0) + (6 x 1)} = 11
in 3. : 2 + {(2 x 4) + (6 x 0)} = 10
in 4. : 1 + {(2 x 4) + (6 x 0)} = 9
We choose alignment 4 because it has the minimum distance.