(last update : 08-11-1999)
Phylogenetics
1. Purpose of phylogenetics :
- With the aid of sequences, it should be possible to find the genealogical ties between organisms. Experience learns that closely related organisms have similar sequences, more distantly related organisms have more dissimilar sequences. One objective is to reconstruct the evolutionary relationship between species.
- An other objective is to estimate the time of divergence between two organisms since they last shared a common ancestor.
2. Disclaimers :
- The theory and practical applications of the different models are not universally accepted.
- With one dataset, different software packages can give different results. Changes in the dataset can also give different results. Therefore it is important to have a good alignment to start with.
- Trees based on an alignment of a gene represent the relationship between genes and this is not necessarily the same relationship as between the whole organisms. If trees are calculated based on different genes from organisms, it is possible that these trees result in different relationships.
3. Terminology :
- node : a node represents a taxonomic unit. This can be a taxon (an existing species) or an ancestor (unknown species : represents the ancestor of 2 or more species).
- branch : defines the relationship between the taxa in terms of descent and ancestry.
- topology : is the branching pattern.
- branch length : often represents the number of changes that have occurred in that branch.
- root : is the common ancestor of all taxa.
- distance scale : scale which represents the number of differences between sequences (e.g. 0.1 means 10 % differences between two sequences)
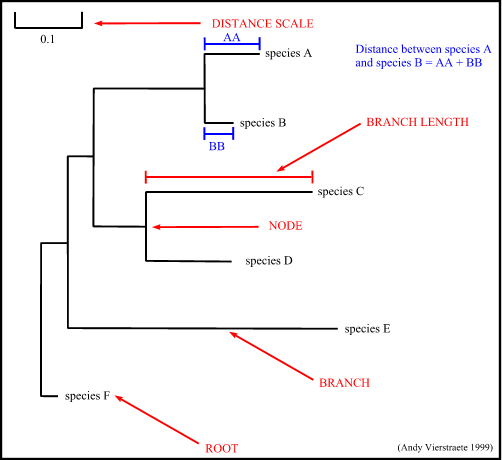
Figure 14 : The tree terminology.
4. Possible ways of drawing a tree :
Trees can be drawn in different ways. There are trees with unscaled branches and with scaled branches.
- Unscaled branches : the length is not proportional to the number of changes. Sometimes, the number of changes are indicated on the branches with numbers. The nodes represents the divergence event on a time scale.
- Scaled branches : the length of the branch is proportional to the number of changes. The distance between 2 species is the sum of the length of all branches connecting them.
Is is also possible to draw these trees with or without a root. For rooted trees, the root is the common ancestor. For each species, there is a unique path that leads from the root to that species. The direction of each path corresponds to evolutionary time. An unrooted tree specifies the relationships among species and does not define the evolutionary path.
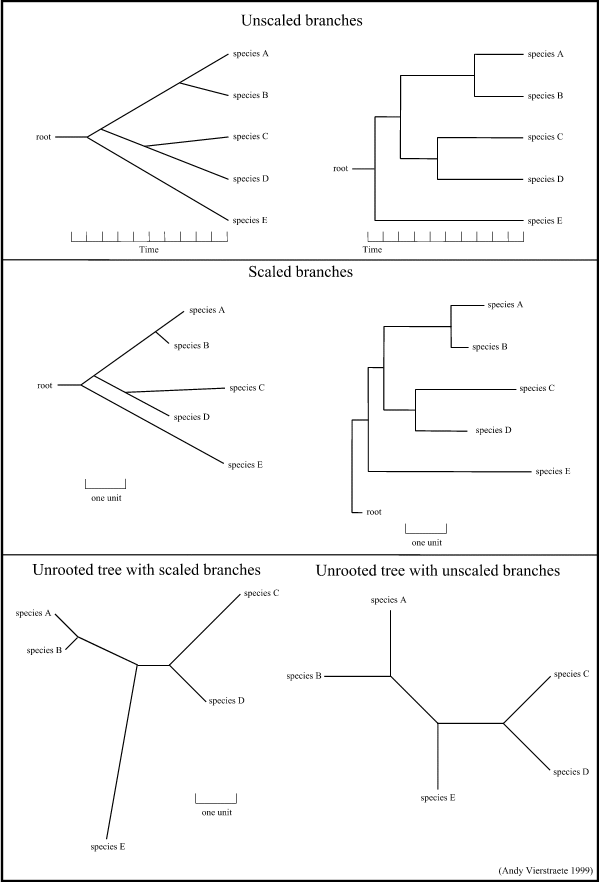
Figure 15 : Some possibilities for drawing a tree. (these are just a few examples, there are a lot of variations possible)
5. Methods of phylogenetic analysis :
There are two major groups of analyses to examine phylogenetic relationships between sequences :
- Phenetic methods : trees are calculated by similarities of sequences and are based on distance methods. The resulting tree is called a dendrogram and does not necessarily reflect evolutionary relationships. Distance methods compress all of the individual differences between pairs of sequences into a single number.
- Cladistic methods : trees are calculated by considering the various possible pathways of evolution and are based on parsimony or likelihood methods. The resulting tree is called a cladogram. Cladistic methods use each alignment position as evolutionary information to build a tree.
5.1. Phenetic methods based on distances :
- Starting from an alignment, pairwise distances are calculated between DNA sequences as the sum of all base pair differences between two sequences (the most similar sequences are assumed to be closely related). This creates a distance matrix.
- All base changes can be considered equally or a matrix of the possible replacements can be used.
- Insertions and deletions are given a larger weight than replacements. Insertions or deletions of multiple bases at one position are given less weight than multiple independent insertions or deletions.
- it is possible to correct for multiple substitutions at a single site.
- From the obtained distance matrix, a phylogenetic tree is calculated with clustering algorithms. These cluster methods construct a tree by linking the least distant pair of taxa, followed by successively more distant taxa.
- UPGMA clustering (Unweighted Pair Group Method using Arithmetic averages) : this is the simplest method
- Neighbor Joining : this method tries to correct the UPGMA method for its assumption that the rate of evolution is the same in all taxa.
5.2. Cladistic methods based on Parsimony :
For each position in the alignment, all possible trees are evaluated and are given a score based on the number of evolutionary changes needed to produce the observed sequence changes. The most parsimonious tree is the one with the fewest evolutionary changes for all sequences to derive from a common ancestor. This is a more time-consuming method than the distance methods.
5.3. Cladistic methods based on Maximum Likelihood :
This method also uses each position in an alignment, evaluates all possible trees, and calculates the likelihood for each tree using an explicit model of evolution (<-> Parsimony just looks for the fewest evolutionary changes). The likelihood's for each aligned position are then multiplied to provide a likelihood for each tree. The tree with the maximum likelihood is the most probable tree. This is the slowest method of all but seems to give the best result and the most information about the tree.
6. Theoretical problems with evolutionary changes between sequences
- Transitions : substitutions from A to G ; G to A ; C to T ; T to C.
- Transversions : substitutions from G to C ; C to G ; T to A ; A to T.
- Deletions : removal of one or more nucleotides.
- Insertion : addition of one or more nucleotides.
- Inversion : rotation of 180 °C of a double stranded DNA segment compromised of of 2 or more base pairs.
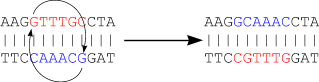
The next figure shows that there is a chance that many more mutations occur than visible at a certain time. Even the best evolutionary models can't solve this problem...
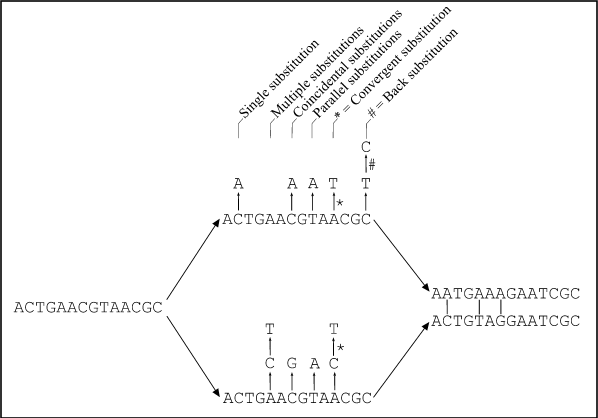
Figure : Two homologous DNA sequences which descended from an ancestral sequence and accumulated mutations since their divergence from each other. Note that although 12 mutations have accumulated, differences can be detected at only three nucleotide sites. (from Fundamentals of Molecular Evolution, Wen-Hsiung Li and Dan Graur, 1991)
back to homepage